Image Credit: University of North Carolina at Charlotte.
However, until recently, the technology to get high-resolution genomic data did not exist, and there were no dependable means to assure that the data was of high quality and utility.
Since the Human Genome Project in the 1990s, scientists have been examining genomes to understand the secrets of life, from discovering particular genes related to sickle cell anemia and breast cancer to developing mRNA vaccines in the ongoing COVID-19 pandemic.
Technology has advanced since those early stages of grouping thousands of cells together to decrypt the millions of base pairs that make up genetic information, and in 2009, researchers developed scRNA-Seq, which only sequences the transcriptome, or the expressed portion of the genome, in a single living cell. This is now widely used in biomedical research.
Unfortunately, scRNA-Seq data is noisy and prone to errors. When a single cell is sequenced rather than a large number of cells, “dropouts”—missing genes in the data—occur often. A single cell, like a single human cell, may have its health difficulties or be at an inconvenient point of its life cycle—it may have just split or be on the edge of cell death—which might lead to additional errors or technical differences in the scRNA-Seq data.
Aside from single-cell concerns, genomic profiling is frequently associated with “normal” sequencing errors. Before the data can be utilized or comprehended, all of these errors must be “cleaned” from it, which is where the new AI algorithm comes in.
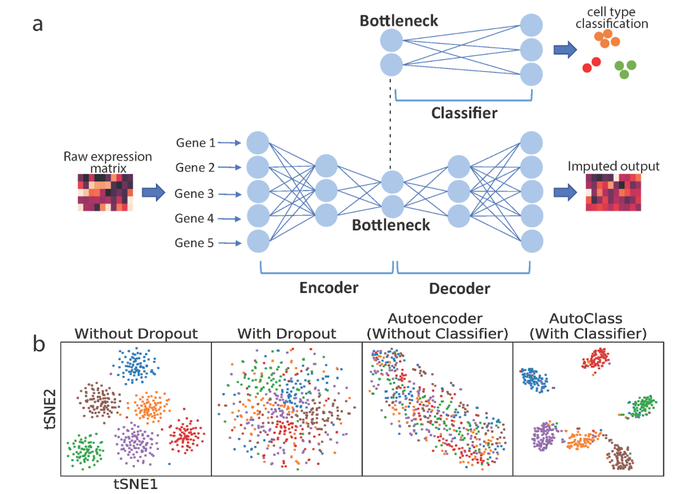
The AutoClass technique is a step forward from previous statistical approaches. Most existing approaches presume that errors (or noises) will follow a predetermined distribution, indicating how frequently they will occur and how large they can be.
Existing approaches are frequently unable to adequately clean data to disclose biological signals, and may even introduce new errors as a result of incorrect data distribution assumptions. AutoClass, on the other hand, has no distributional assumptions and, as a result, can efficiently remedy a wide range of noises or technological variances.
AutoClass is an AI algorithm based on a special deep neural network designed to maximize both noise removal and signal retention. The AI teaches itself to differentiate signal vs noise in the data by seeing enough data. Usually the more data it sees, the better it performs.
Dr. Weijun Luo, Department of Bioinformatics and Genomics, College of Computing and Informatics, University of North Carolina at Charlotte
Dr. Luo and his colleagues demonstrated in the study that AutoClass can rebuild high-quality scRNA-Seq data and improve downstream analysis in a variety of ways. AutoClass is also robust, performing well in a variety of scRNA-Seq data formats and situations.
AutoClass is very efficient and scalable, and it works well with data of various sample sizes and feature sizes. It also operates smoothly on a regular PC or laptop.
Journal Reference:
Li, H., et al. (2022) A universal deep neural network for in-depth cleaning of single-cell RNA-Seq data. Nature Communications. doi.org/10.1038/s41467-022-29576-y.